polars-bio 0.31.0: VCF Zarr Support for Array-Native Variant Analytics
polars-bio 0.31.0 adds first-class support for VCF Zarr, giving Python users a fast, lazy, DataFusion-backed way to query VCF-derived Zarr stores with Polars.
polars-bio 0.31.0 adds first-class support for VCF Zarr, giving Python users a fast, lazy, DataFusion-backed way to query VCF-derived Zarr stores with Polars.
polars-bio 0.29.0 adds support for Pandas >= 3.0.0. Since pandas 3.0 made PyArrow-backed data even more central, with the new default string dtype using pyarrow under the hood when available, we wanted to measure what that means for interval workloads in practice.
So instead of comparing different interval libraries, this benchmark compares different input and execution paths through the same polars-bio range engine:
DataFrameDataFrameLazyFrameThe question is simple: how much overhead do you pay once data is materialized into a Python DataFrame, and how much of that gap can Arrow-backed Pandas close?
polars-bio 0.26.0 brings first-class GTF format support, automatic SAM tag type inference for custom/nanopore tags, and critical bug fixes for multi-partition writes and VCF contig metadata.
Back in September 2025 we benchmarked three libraries across three operations. A lot has changed since then. In December 2025, pyranges1 published a preprint describing its Rust-powered backend (ruranges) and an expanded set of interval operations. On the polars-bio side, version 0.24.0 ships a fully rewritten range-operations engine built on upstream DataFusion UDTF providers (OverlapProvider, NearestProvider, and the new coverage/cluster/complement/merge/subtract providers from datafusion-bio-function-ranges), replacing the earlier sequila-native backend.
Genomic analyses in Python typically start with reading BAM, VCF, or FASTQ files into memory. The choice of library for this step can have a dramatic impact on both wall-clock time and memory consumption — especially as datasets grow to tens or hundreds of millions of records.
pysam has long been the go-to Python library for working with these formats. It provides comprehensive bindings to htslib and is battle-tested across thousands of projects. However, several newer libraries have emerged that leverage Apache Arrow columnar format and Rust-based parsers to offer potentially better performance.
In this post, we benchmark four Python libraries head-to-head on real-world genomic data to find out which offers the best combination of speed and memory efficiency for reading BAM, VCF, and FASTQ files.
polars-bio 0.23.0 is here with significant parsing performance improvements across VCF, BAM, and FASTQ formats, plus first-day support for Python 3.14. This release bumps the underlying datafusion-bio-formats engine to 0.5.0, delivering up to 3.6x faster VCF parsing with no API changes required.
We're excited to announce significant performance improvements to GFF file reading in polars-bio 0.15.0. This release introduces two major optimizations that dramatically improve both speed and memory efficiency when working with GFF files:
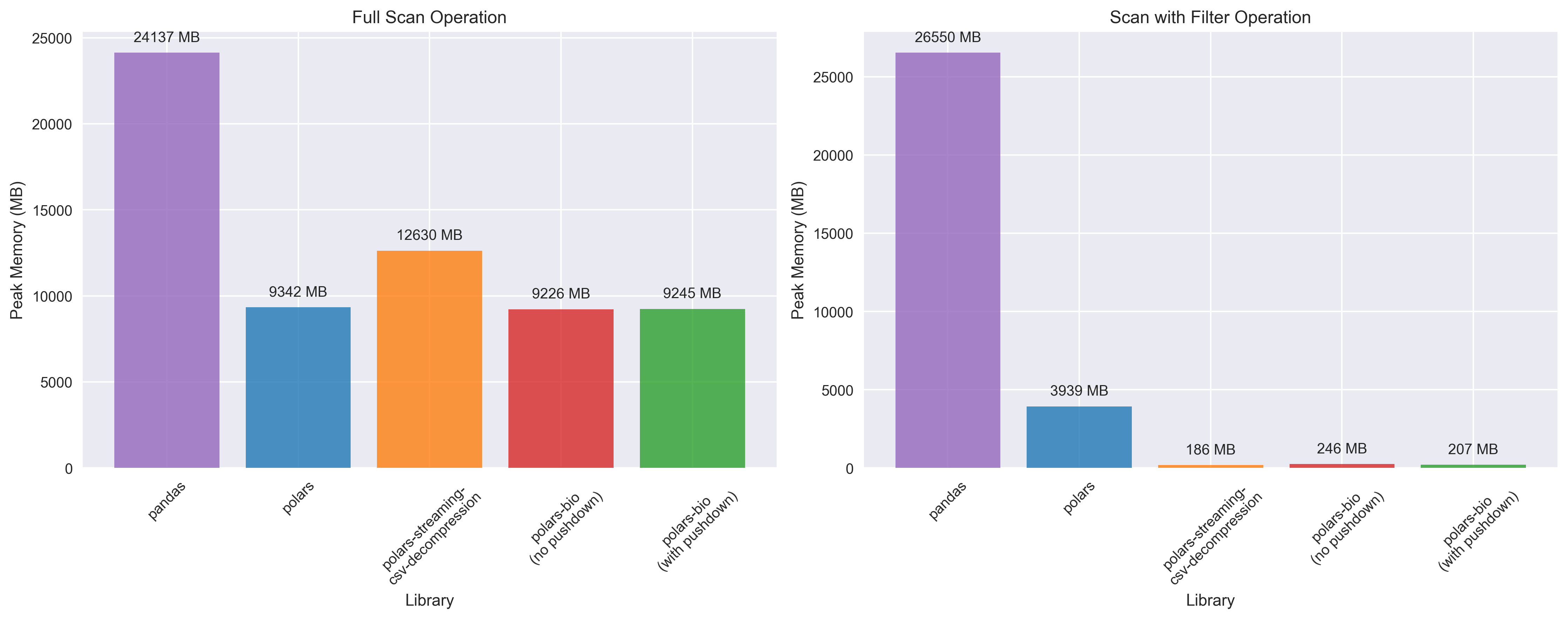
Projection Pushdown: Only the columns you need are read from disk, reducing I/O overhead and memory usage. This is particularly beneficial when working with wide GFF files that contain many optional attributes.
Predicate Pushdown: Row filtering is applied during the file reading process, eliminating the need to load irrelevant data into memory. This allows for lightning-fast queries on large GFF datasets.
Fully Streamed Parallel Reads: BGZF-compressed files can now be read in parallel with true streaming, enabling out-of-core processing of massive genomic datasets without memory constraints.
To evaluate these improvements, we conducted comprehensive benchmarks comparing three popular data processing libraries:
All benchmarks were performed on a large GFF file (~7.7 million records, file and index needed for parallel reading) with both full scan and filtered query scenarios to demonstrate real-world performance gains.
For pandas and polars reading, we used the following methods (thanks to @urineri for the Polars code).
Since Polars decompresses compressed CSV/TSV files completely in memory as highlighted here, we also used polars_streaming_csv_decompression, a great plugin developed by @ghuls to enable streaming decompression in Polars.
Test query used for filtered benchmarks (Polars and polars-bio):
result = (
lf.filter(
(pl.col("seqid") == "chrY")
& (pl.col("start") < 500000)
& (pl.col("end") > 510000)
)
.select(["seqid", "start", "end", "type"])
.collect()
)
The above query is very selective and returns only two rows from the entire dataset.
Complete benchmark code and results are available in the polars-bio repository.
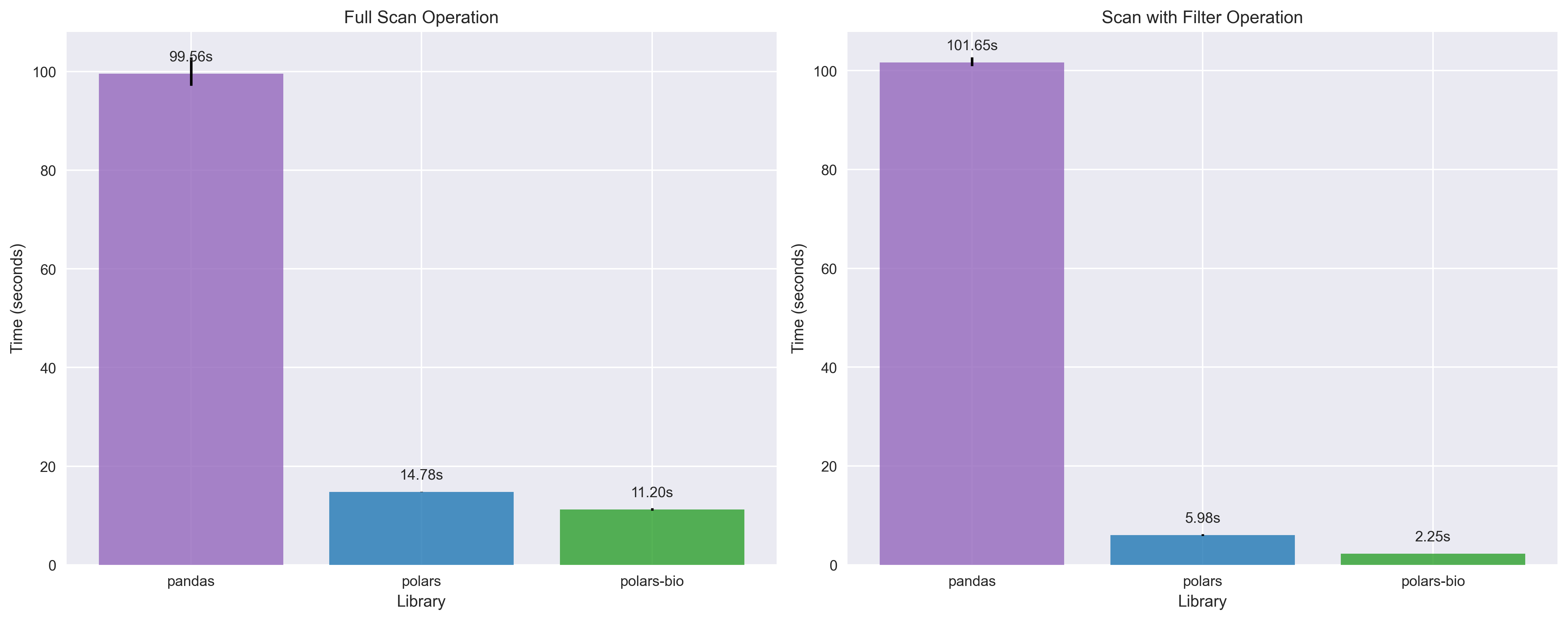
Key takeaways:
Key takeaways:
polars_streaming_csv_decompression can use more than 20x less memory than vanilla Polars and more than two orders of magnitude less memory than Pandas for operations involving filtering.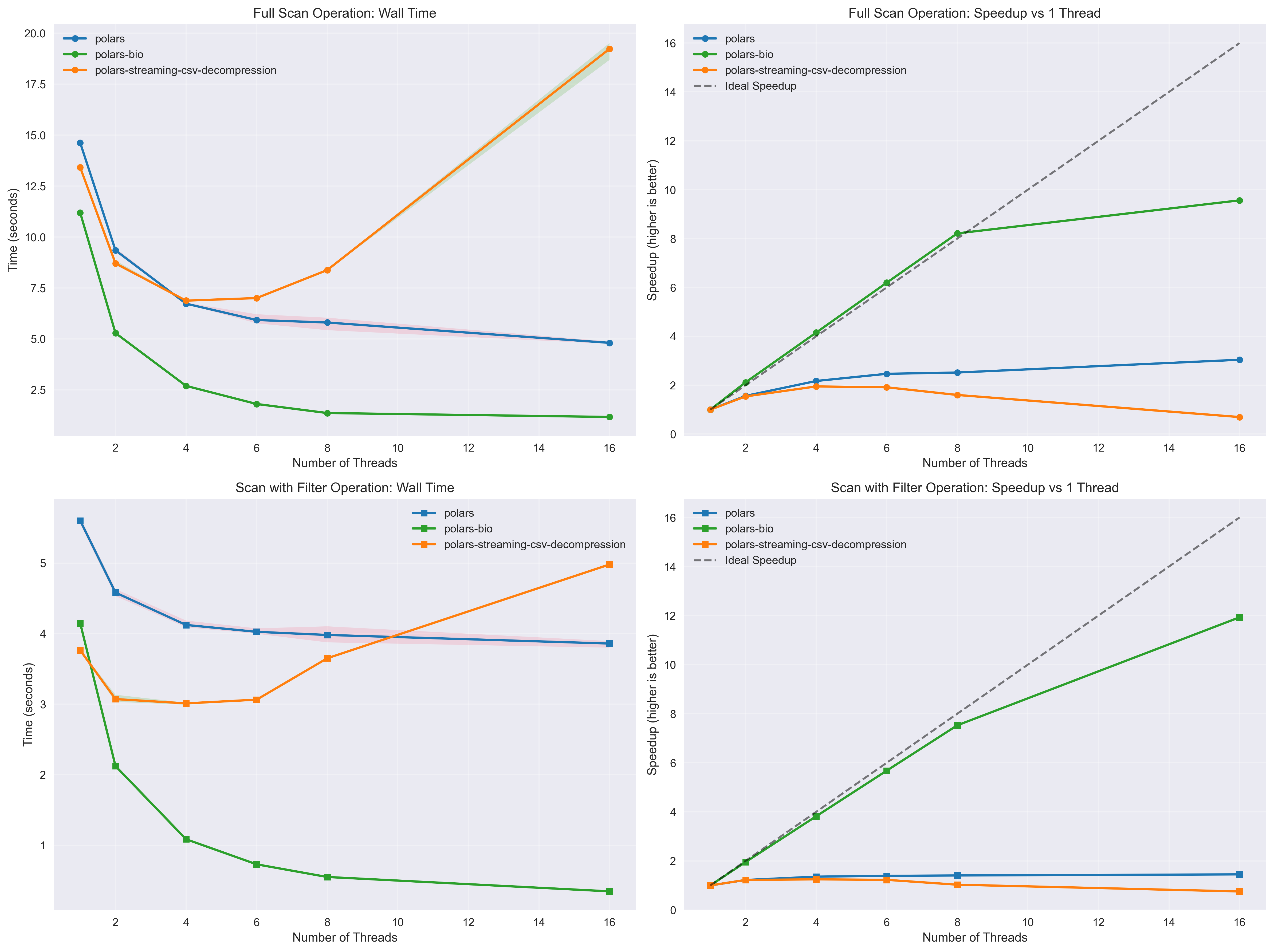
Key takeaways:
The benchmarks demonstrate that polars-bio 0.15.0 delivers significant performance improvements for GFF file processing. These optimizations, combined with near-linear thread scaling and fully streamed parallel reads, make polars-bio an ideal choice for high-performance genomic data analysis workflows.
If you haven't tried polars-bio yet, now is a great time to explore its capabilities for efficient genomic data processing with Python! Join our upcoming seminar on September 15, 2025, to learn more about polars-bio and its applications in genomics.

Benchmarking isn’t a one-and-done exercise—it’s a moving target. As tools evolve, new versions can shift performance profiles in meaningful ways, so keeping results current is just as important as the first round of measurements.
Recently, three novel libraries that have started to gain traction: pyranges1, GenomicRanges and polars-bio
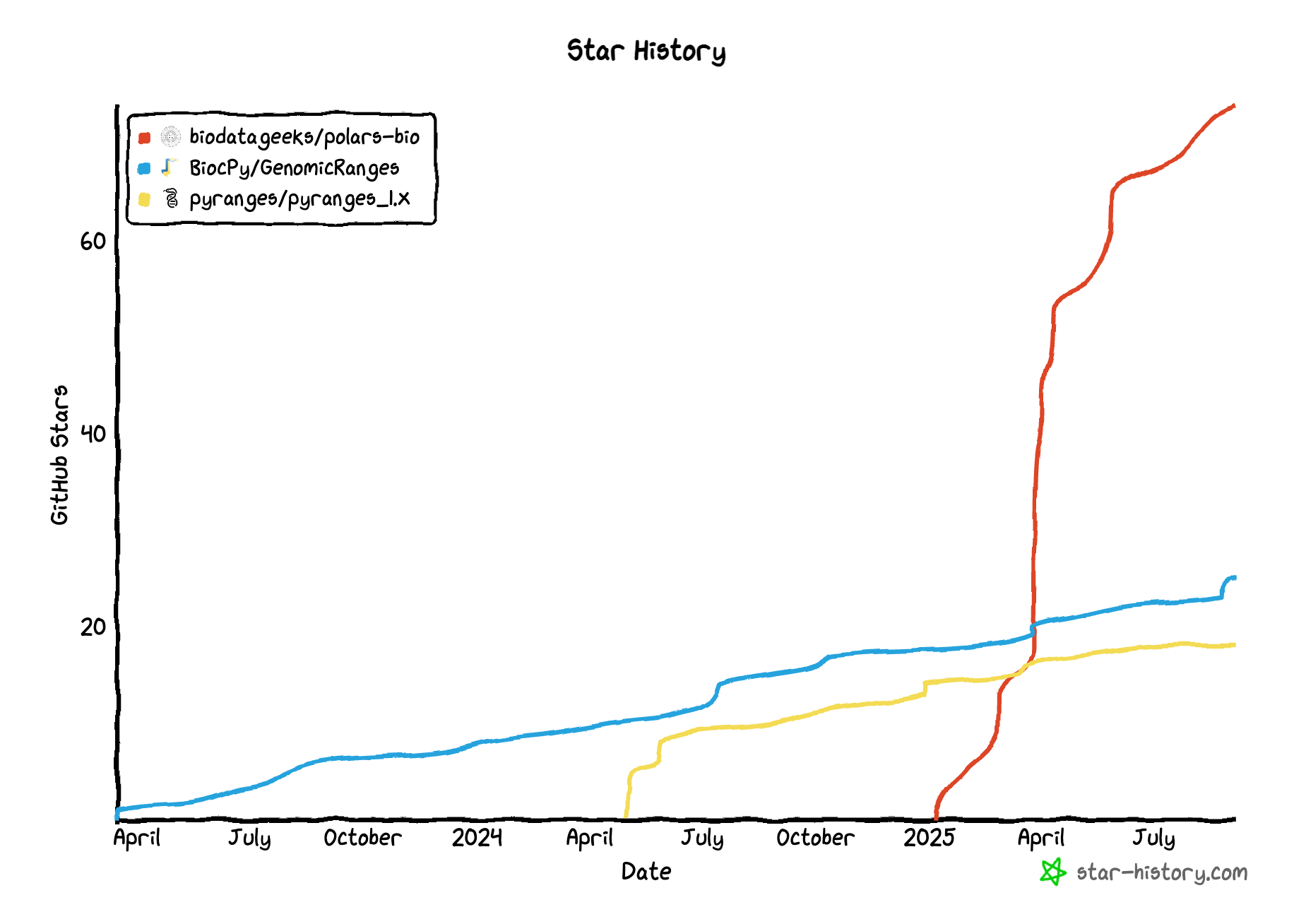
shipped major updates:
0.12.0 it supports:
Each of these changes has the potential to meaningfully alter performance and memory characteristics for common genomic interval tasks.
In this post, we revisit our benchmarks with those releases in mind. We focus on three everyday operations:
For comparability, we use the same AIList dataset from our previous write-up, so you can see exactly how the new backends and data structures change the picture. Let’s dive in and see what’s faster, what’s leaner, and where the trade-offs now live.
| Dataset pairs | Size | # of overlaps (1-based) |
|---|---|---|
| 1-2 & 2-1 | Small | 54,246 |
| 7-3 & 3-7 | Medium | 4,408,383 |
| 8-7 & 7-8 | Large | 307,184,634 |
| Library | Version |
|---|---|
| polars_bio | 0.13.1 |
| pyranges | 0.1.14 |
| genomicranges | 0.7.2 |
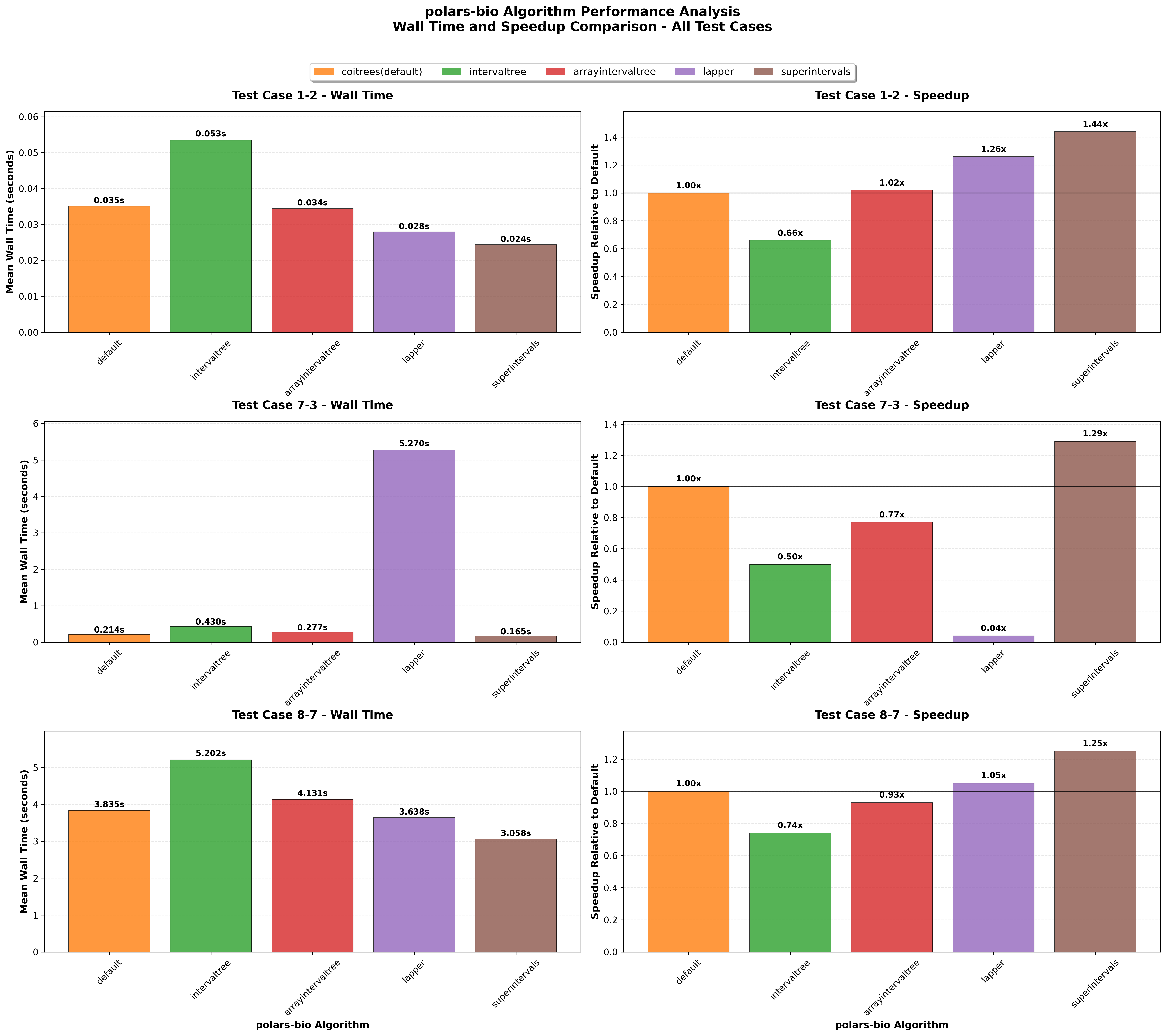
Key takeaways:
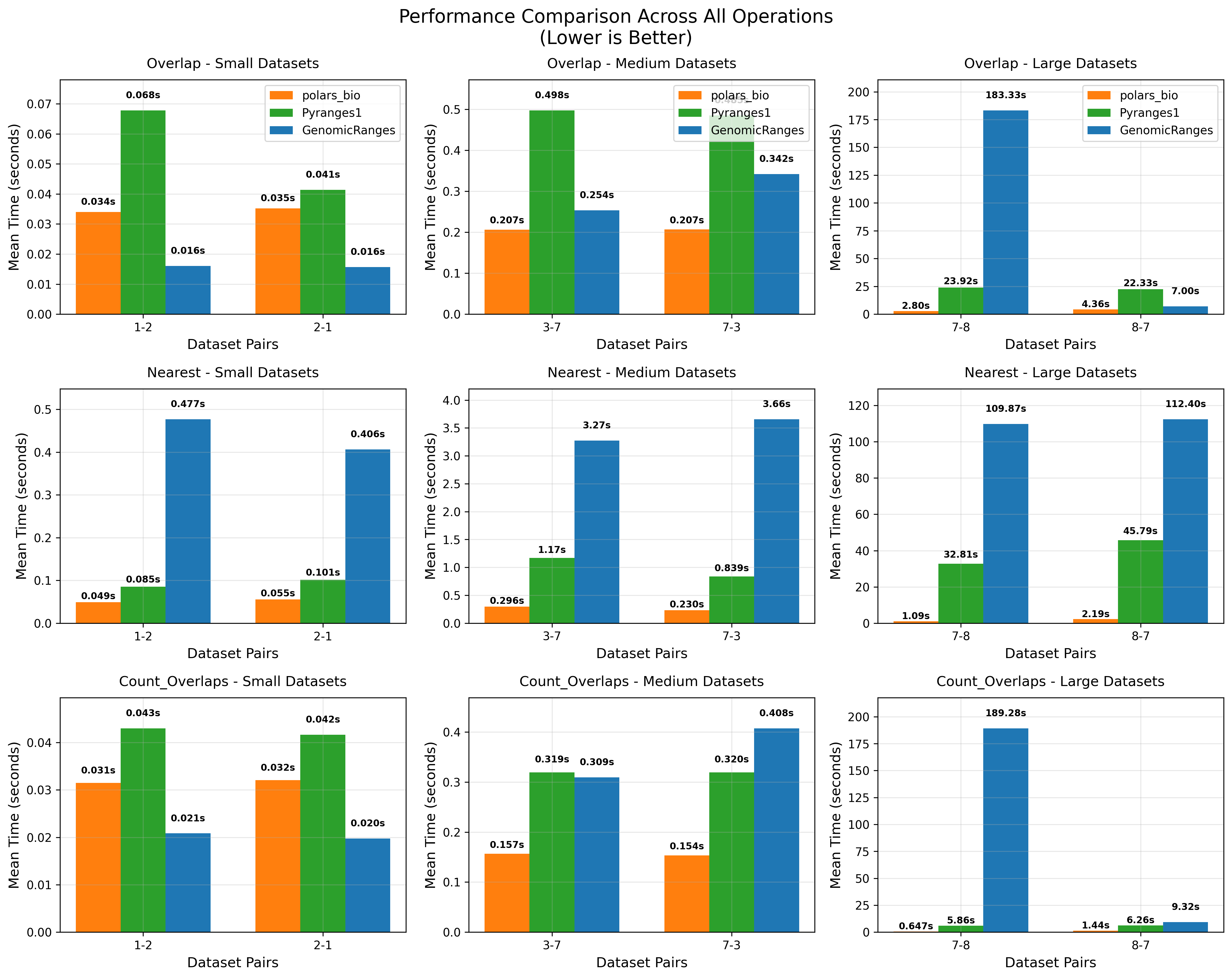
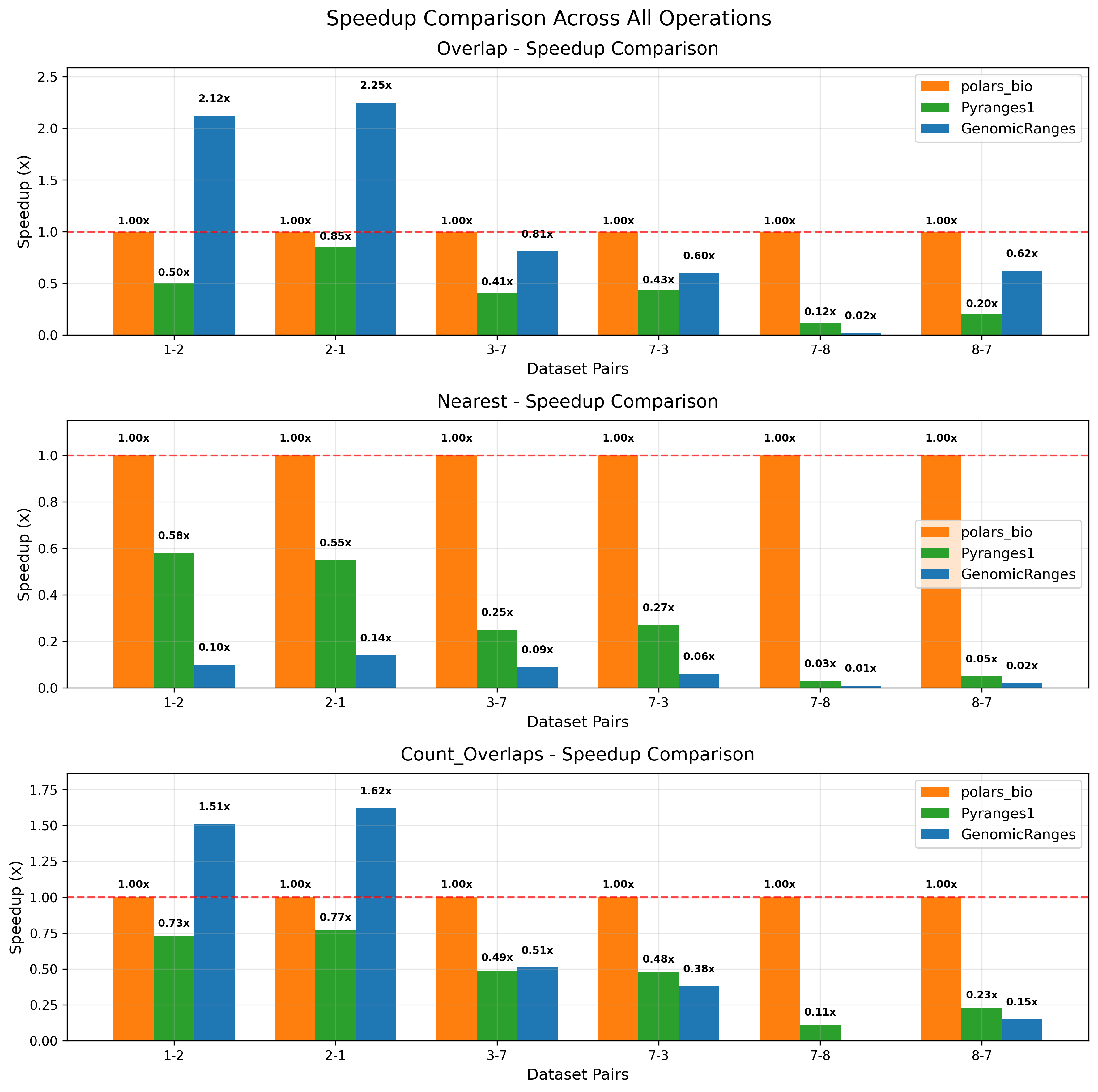
Key takeaways:
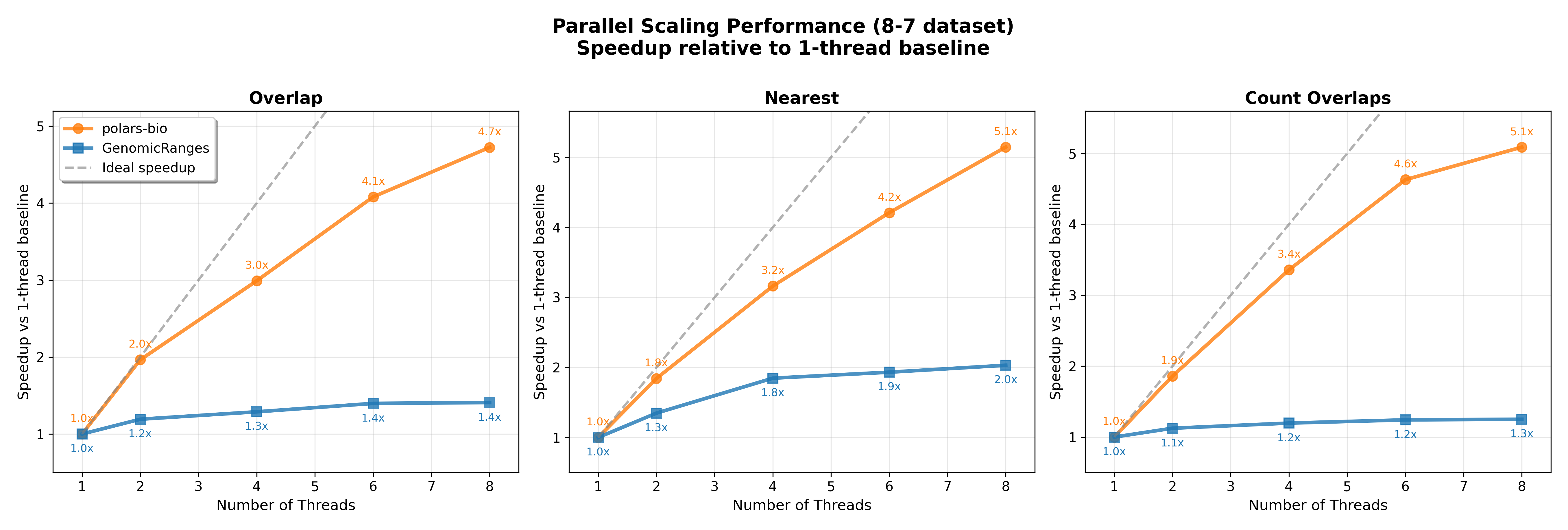
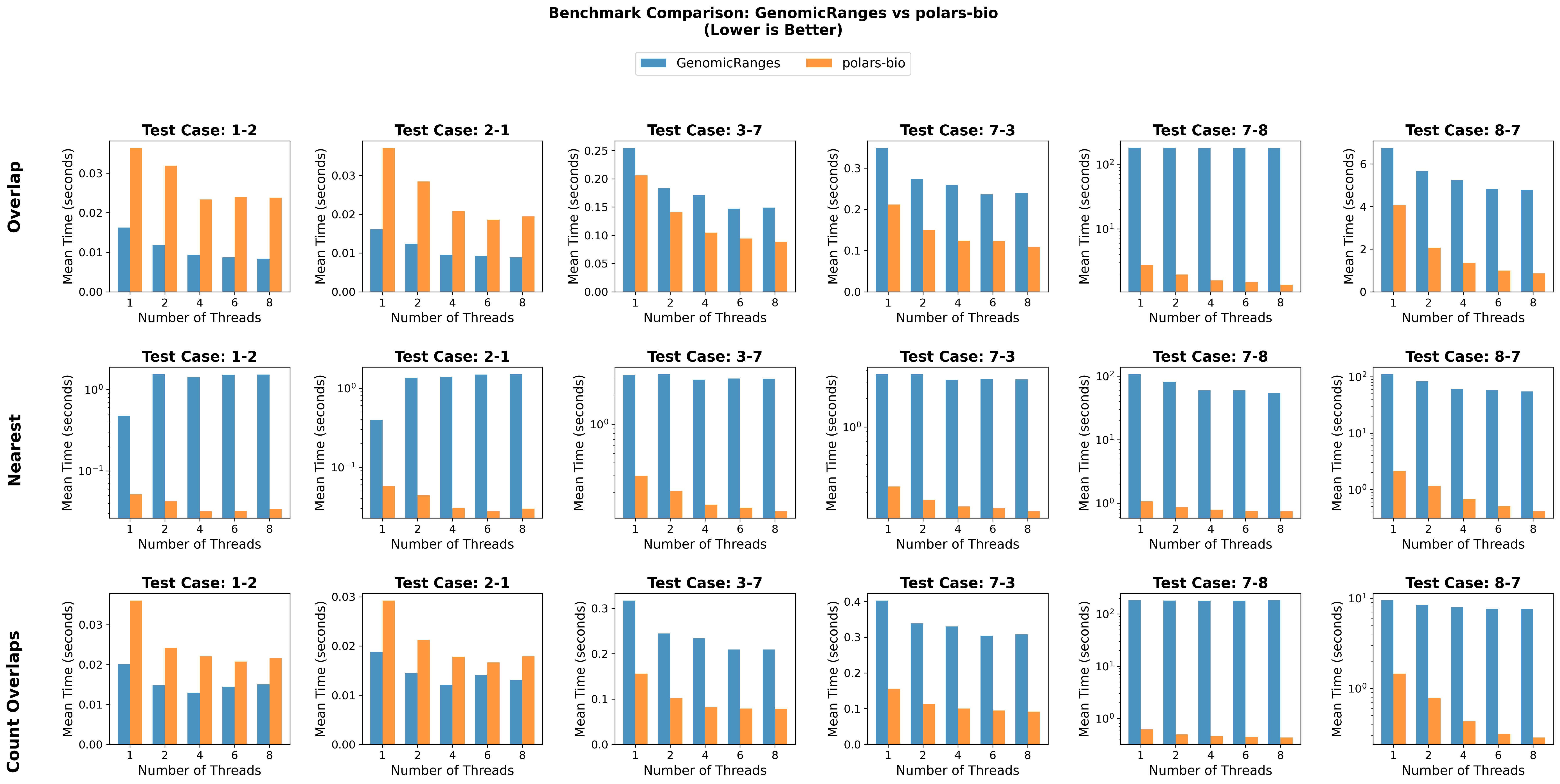
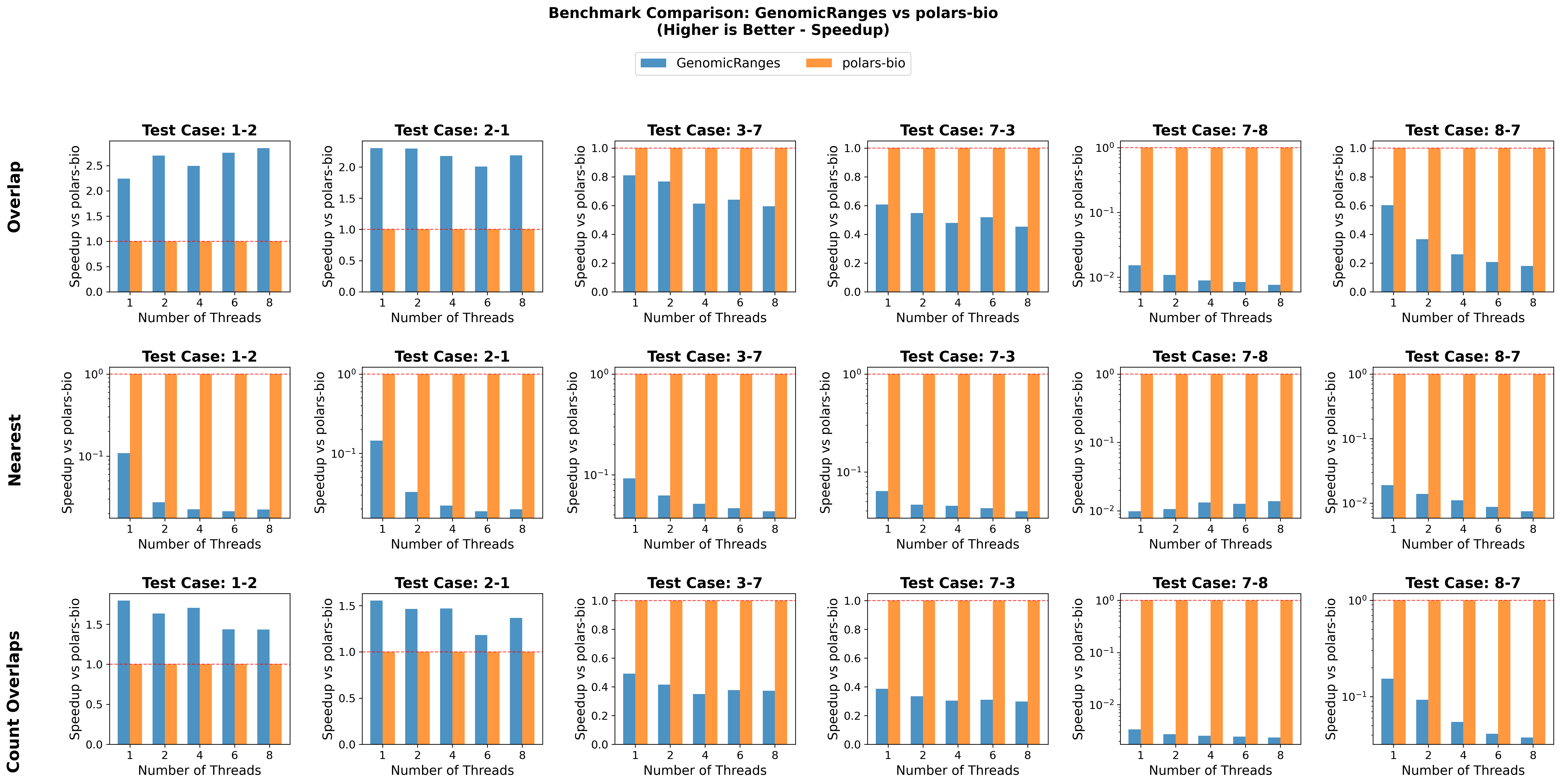
Key takeaways:
Here we compare end-to-end performance including data loading, overlap operation, and saving results to CSV.
Info
POLARS_MAX_THREADS=1 was set to ensure fair comparison with single-threaded PyRanges.full rows, code) for fair comparison.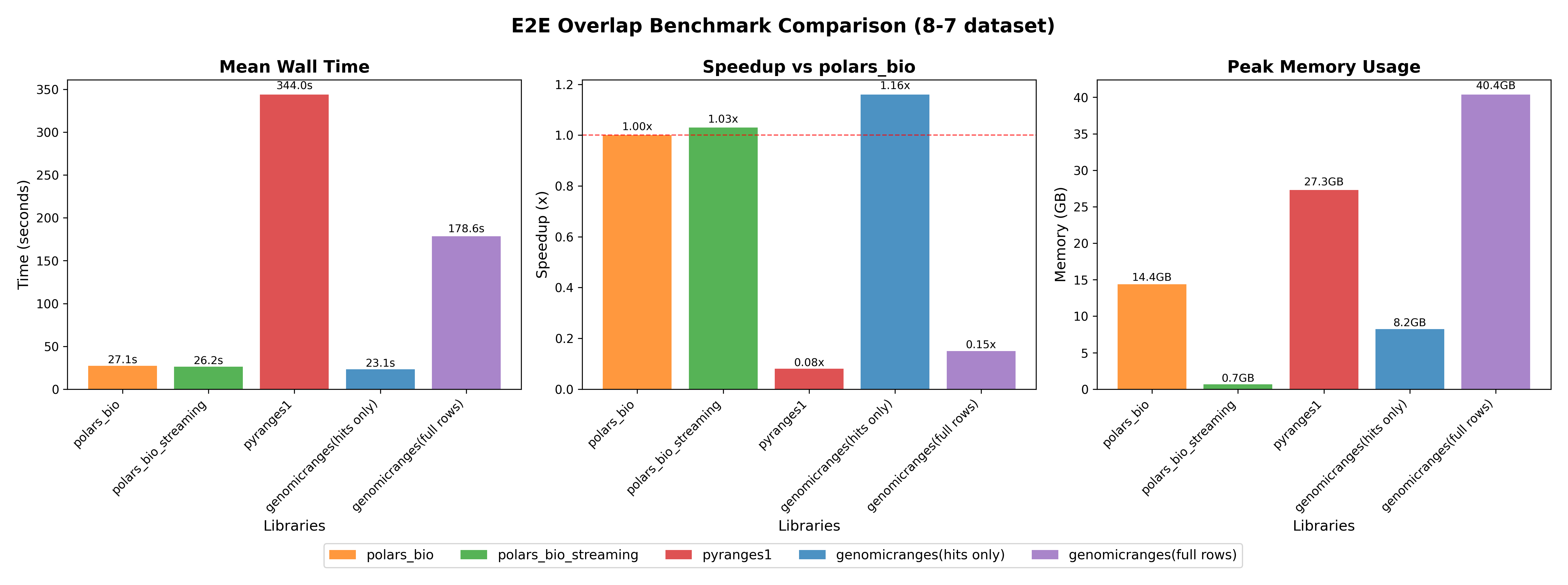
Key takeaways:
For small and medium datasets, all tools perform well; at large scale, polars-bio excels with better scalability and memory efficiency, achieving an ultra‑low footprint in streaming mode.