Benchmarks
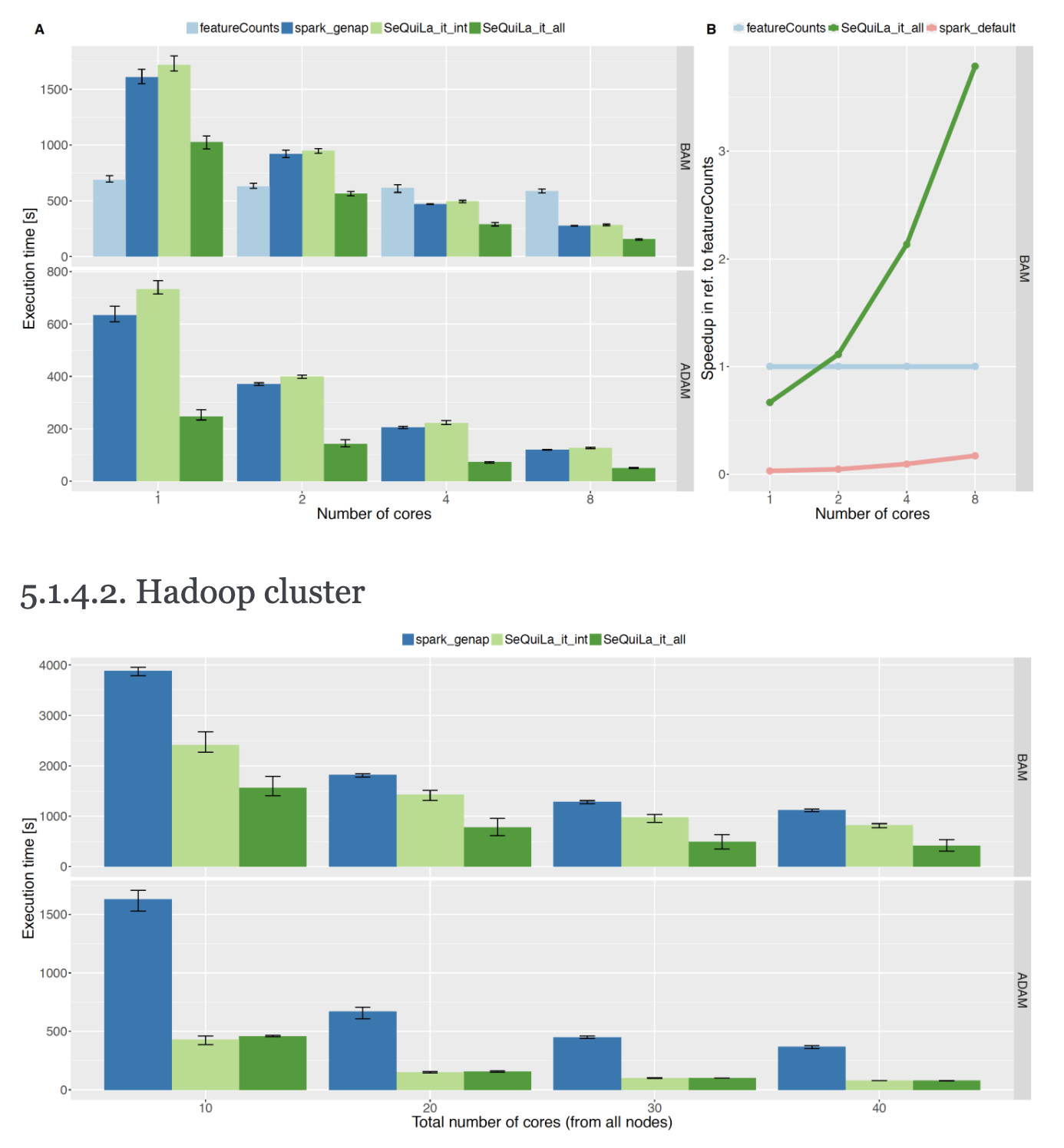
Interval joins
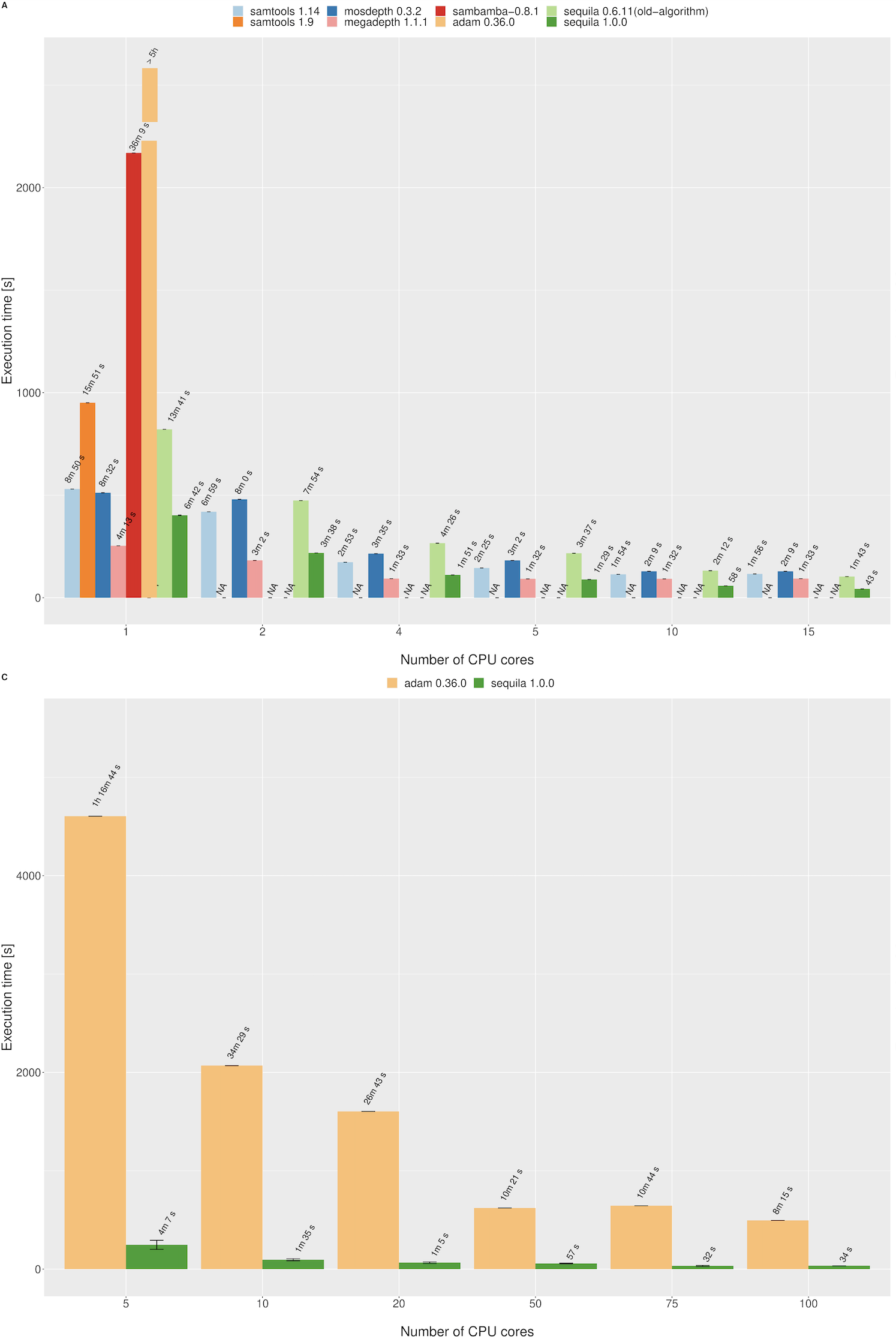
Coverage
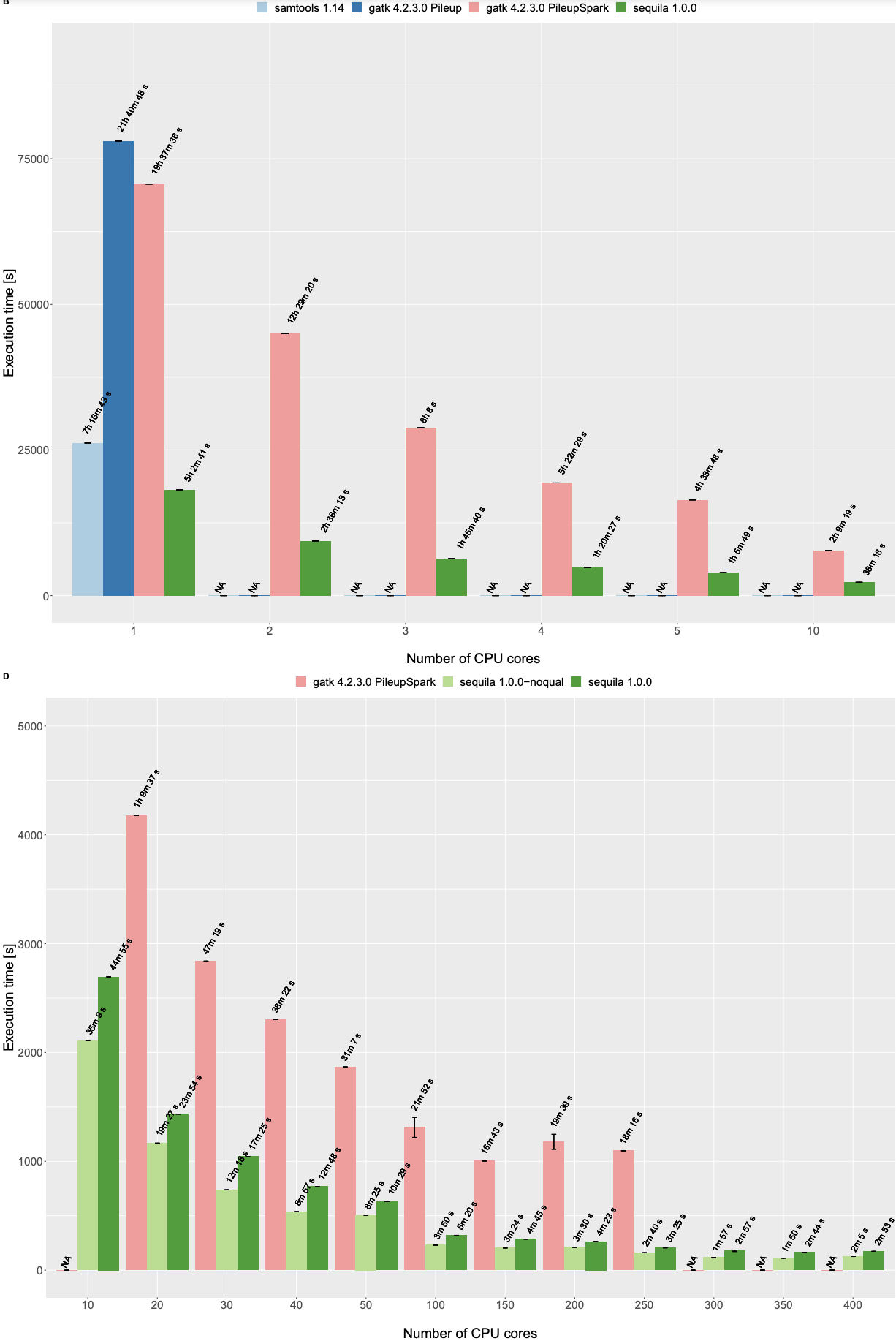
Pileup
Getting started
# ensure you have Apache Spark and GraalVM installed
sdk install spark 3.2.2
sdk install java 21.3.0.r11-grl
# set Apache Spark and JDK using sdkman
sdk use spark 3.2.2
# use GraalVM for best performance
sdk use java 21.3.0.r11-grl
# in case you prefer to use a Python interface
pip install pysequila==0.4.0
# download sample data
mkdir -p data
#BAM
wget -nc https://github.com/biodatageeks/pysequila/raw/master/features/data/NA12878.multichrom.md.bam -O data/NA12878.bam
wget -nc https://github.com/biodatageeks/pysequila/raw/master/features/data/NA12878.multichrom.md.bam.bai -O data/NA12878.bam.bai
#Reference
wget -nc https://github.com/biodatageeks/pysequila/raw/master/features/data/Homo_sapiens_assembly18_chr1_chrM.small.fasta -O data/hg18.fasta
wget -nc https://github.com/biodatageeks/pysequila/raw/master/features/data/Homo_sapiens_assembly18_chr1_chrM.small.fasta.fai -O data/hg18.fasta.fai
# targets
wget -nc https://github.com/biodatageeks/pysequila/raw/master/features/data/targets.bed -O data/targets.bed
-- remember to initialize SeQuiLaSession in Python or Scala REPL first !
-- create a table over a BAM file
-- replace {{ PWD }} with your location
CREATE TABLE IF NOT EXISTS reads
USING org.biodatageeks.sequila.datasources.BAM.BAMDataSource
OPTIONS(path "{{ PWD }}/data/NA12878.bam");
-- calculate pileup
SELECT * FROM pileup('reads', 'NA12878','{{ PWD }}/data/hg18.fasta', false) LIMIT 5;
+------+---------+-------+---------+--------+--------+-----------+----+-----+
|contig|pos_start|pos_end| ref|coverage|countRef|countNonRef|alts|quals|
+------+---------+-------+---------+--------+--------+-----------+----+-----+
| 1| 34| 34| C| 1| 1| 0|null| null|
| 1| 35| 35| C| 2| 2| 0|null| null|
| 1| 36| 37| CT| 3| 3| 0|null| null|
| 1| 38| 40| AAC| 4| 4| 0|null| null|
| 1| 41| 49|CCTAACCCT| 5| 5| 0|null| null|
+------+---------+-------+---------+--------+--------+-----------+----+-----+
only showing top 5 rows
pyspark --master local[1] \
--driver-memory 4g \
--packages org.biodatageeks:sequila_2.12:1.1.1
from pysequila import SequilaSession
# initialize an extended SparkSession
ss = SequilaSession \
.builder \
.getOrCreate()
# calculate pileup
# replace {{ PWD }} with your location
ss.pileup("{{ PWD }}/data/NA12878.bam", "{{ PWD }}/data/hg18.fasta", False).show(5)
+------+---------+-------+---------+--------+--------+-----------+----+-----+
|contig|pos_start|pos_end| ref|coverage|countRef|countNonRef|alts|quals|
+------+---------+-------+---------+--------+--------+-----------+----+-----+
| 1| 34| 34| C| 1| 1| 0|null| null|
| 1| 35| 35| C| 2| 2| 0|null| null|
| 1| 36| 37| CT| 3| 3| 0|null| null|
| 1| 38| 40| AAC| 4| 4| 0|null| null|
| 1| 41| 49|CCTAACCCT| 5| 5| 0|null| null|
+------+---------+-------+---------+--------+--------+-----------+----+-----+
only showing top 5 rows
-- remember to initialize SeQuiLaSession in Python or Scala REPL first !
-- create a table over a BAM file
CREATE TABLE IF NOT EXISTS reads
USING org.biodatageeks.sequila.datasources.BAM.BAMDataSource
OPTIONS(path "{{ PWD }}/data/NA12878.bam");
-- calculate coverage
SELECT * FROM coverage('reads', 'NA12878','{{ PWD }}/data/hg18.fasta') LIMIT 10;
+------+---------+-------+---+--------+
|contig|pos_start|pos_end|ref|coverage|
+------+---------+-------+---+--------+
| 1| 34| 34| R| 1|
| 1| 35| 35| R| 2|
| 1| 36| 37| R| 3|
| 1| 38| 40| R| 4|
| 1| 41| 49| R| 5|
| 1| 50| 67| R| 6|
| 1| 68| 109| R| 7|
| 1| 110| 110| R| 6|
| 1| 111| 111| R| 5|
| 1| 112| 113| R| 4|
+------+---------+-------+---+--------+
only showing top 10 rows
pyspark --master local[1] \
--driver-memory 4g \
--packages org.biodatageeks:sequila_2.12:1.1.1
from pysequila import SequilaSession
# initialize an extended SparkSession
ss = SequilaSession \
.builder \
.getOrCreate()
# calculate pileup
# replace {{ PWD }} with your location
ss.coverage("{{ PWD }}/data/NA12878.bam", "{{ PWD }}/data/hg18.fasta").show(10)
+------+---------+-------+---+--------+
|contig|pos_start|pos_end|ref|coverage|
+------+---------+-------+---+--------+
| 1| 34| 34| R| 1|
| 1| 35| 35| R| 2|
| 1| 36| 37| R| 3|
| 1| 38| 40| R| 4|
| 1| 41| 49| R| 5|
| 1| 50| 67| R| 6|
| 1| 68| 109| R| 7|
| 1| 110| 110| R| 6|
| 1| 111| 111| R| 5|
| 1| 112| 113| R| 4|
+------+---------+-------+---+--------+
only showing top 10 rows
-- remember to initialize SeQuiLaSession in Python or Scala REPL first !
-- create a table over a BED file
CREATE TABLE IF NOT EXISTS targets
USING org.biodatageeks.sequila.datasources.BED.BEDDataSource
OPTIONS(path "{{ PWD }}/data/targets.bed")
-- create a table over a BAM file
CREATE TABLE IF NOT EXISTS reads
USING org.biodatageeks.sequila.datasources.BAM.BAMDataSource
OPTIONS(path "{{ PWD }}/data/NA12878.bam");
-- interval join
EXPLAIN SELECT distinct reads.contig FROM reads JOIN targets ON
(
targets.contig=reads.contig
AND
reads.pos_end >= targets.pos_start
AND
reads.pos_start <= targets.pos_end
)
1
2
3
4
5
6
7
8
9
10
11
|== Physical Plan ==
*(4) HashAggregate(keys=[contig#538], functions=[])
+- Exchange hashpartitioning(contig#538, 200), ENSURE_REQUIREMENTS, [id=#167]
+- *(3) HashAggregate(keys=[contig#538], functions=[])
+- *(3) Project [contig#538]
+- IntervalTreeJoinOptimChromosome [pos_start#540, pos_end#541, pos_start#638, pos_end#639, contig#538, contig#637], SequilaSession(org.apache.spark.sql.SparkSession@1323e240), 1, 0, false, org.biodatageeks.sequila.rangejoins.methods.IntervalTree.IntervalTreeRedBlack
:- *(1) Filter isnotnull(contig#538)
: +- *(1) Scan org.biodatageeks.sequila.datasources.BAM.BDGAlignmentRelation@2e108b3e default.reads[contig#538,pos_start#540,pos_end#541] PushedFilters: [], ReadSchema: struct<contig:string,pos_start:int,pos_end:int>
+- *(2) Filter isnotnull(contig#637)
+- *(2) Scan org.biodatageeks.sequila.datasources.BED.BEDRelation@10ee96c5 default.targets[contig#637,pos_start#638,pos_end#639] PushedFilters: [], ReadSchema: struct<contig:string,pos_start:int,pos_end:int>
pyspark --master local[1] \
--driver-memory 4g \
--packages org.biodatageeks:sequila_2.12:1.1.0
targets_df = ss.read\
.format("org.biodatageeks.sequila.datasources.BED.BEDDataSource")\
.load("{{ PWD }}/data/targets.bed")
reads_df=ss.read\
.format("org.biodatageeks.sequila.datasources.BAM.BAMDataSource")\
.load("/{{ PWD }}/data/NA12878.bam")
reads_df.join(targets_df,\
(reads_df.contig == targets_df.contig)\
& (reads_df.pos_end >= targets_df.pos_start)\
& (reads_df.pos_start <= targets_df.pos_end),\
).select(reads_df.contig)\
.distinct()\
.explain()
1
2
3
4
5
6
7
8
9
10
11
== Physical Plan ==
*(4) HashAggregate(keys=[contig#1096], functions=[])
+- Exchange hashpartitioning(contig#1096, 200), ENSURE_REQUIREMENTS, [id=#267]
+- *(3) HashAggregate(keys=[contig#1096], functions=[])
+- *(3) Project [contig#1096]
+- IntervalTreeJoinOptimChromosome [pos_start#1098, pos_end#1099, pos_start#816, pos_end#817, contig#1096, contig#815], SequilaSession(org.apache.spark.sql.SparkSession@1323e240), 1, 0, false, org.biodatageeks.sequila.rangejoins.methods.IntervalTree.IntervalTreeRedBlack
:- *(1) Filter isnotnull(contig#1096)
: +- *(1) Scan org.biodatageeks.sequila.datasources.BAM.BDGAlignmentRelation@3ae2bfd0 [contig#1096,pos_start#1098,pos_end#1099] PushedFilters: [], ReadSchema: struct<contig:string,pos_start:int,pos_end:int>
+- *(2) Filter isnotnull(contig#815)
+- *(2) Scan org.biodatageeks.sequila.datasources.BED.BEDRelation@502849e0 [contig#815,pos_start#816,pos_end#817] PushedFilters: [], ReadSchema: struct<contig:string,pos_start:int,pos_end:int>
It combines analytical power of Python with SQL syntax for almost unlimited querying and processing of NGS data.
Big data ready, cloud-native
Contributions welcome!
We do a Pull Request contributions workflow on GitHub. New users are always welcome!